Ethnobotanical Leaflets 13:639-47, 2009.
Adenosine deaminase from Plasmodium falciparum as a Potential Drug Target in Anti- Malarial Drug Designing: A Bioinformatic Approach
S. Sriram*, J. Kavitha Srilakshmi, V. Meenaa and C. Sasikumar
PG Department of Biotechnology, Nehru Memorial College (Autonomous), Puthanampatti, Tiruchirappalli
*Corresponding author : Email:
Issued 01 May 2009
Absract
Parasites are responsible for a wide variety of infectious diseases causing an enormous health and economical blight. Malaria is one such prominent disease that causes widespread infections in humans and results in innumerable deaths annually. The development of resistance of the malarial parasites to the conventional drugs has signaled for an urgent need to design new drugs in an effective way and also to identify and study new drug targets to combat this disease. The rational design of a drug is usually based on the biochemical and physiological differences between the pathogen and the host. So in this current study we focus on the striking differences in the purine metabolism of the malarial parasite Plasmodium falciparum and that of the host. Based on this, we submit a hypothesis on targeting a protein Adenosine deaminase that plays an important role in the purine metabolism of the parasite. In this study a synthetic and a natural drug were used and their efficacy was compared and analyzed.
Keywords: Plasmodium falciparum, Adenosine deaminase, Purine pathway.
Background
Malaria (from medieval Italian: mala aria – bad air) formerly called as ague or marsh fever is an infectious disease that causes about 350-500million infections in humans and 1-3million deaths annually. This infectious disease is widespread in many tropical, subtropical regions and mostly prevalent among young children in Sub Saharan Africa [1]. Malaria is caused by a protozoa Plasmodium (Phylum Apicomplexa). In humans malaria is caused by P. falciparum, P. malariae, P. ovale, P. vivax, and P. knowlesi [2][3]. Among the Plasmodium protozoa, Plasmodium falciparum is responsible for about 80% infections and 90% deaths [4]. The parasite’s primary hosts and transmission vectors are female mosquitoes of the genus Anopheles and humans act as intermediates.
The parasite is relatively protected from attack by the body’s immune system because for most of its human life cycle it resides within the liver and blood cells and is relatively invisible to immune surveillance. Even though circulating infected blood cells are destroyed in the spleen, the Plasmodium falciparum, to avoid this fate displays adhesive proteins on the surface of the infected blood cells to stick to the walls of small blood cells, thereby sequestering the parasite from passing through the general circulation and spleen. Although the RBC surface adhesive proteins (PfEMP1) are exposed to the immune system, they do not serve as good immune targets because of their extreme diversity and there are at least 60 different variations of PfEMP1 within a single parasite and limitless variations within parasite population [5]. To compound these problems malarial parasites have become more and more resistant to conventional treatment and drugs.
The present study is about a novel method of drug designing and identifying a drug target based on the differences in the purine pathway of the parasite and host. Unlike their mammalian host most parasites lack the pathways for de novo purine biosysnthesis and rely on the salvage pathway to meet their purine demands [6]. In some cases there are sufficient distinctions between corresponding enzymes of the purine salvage pathway of the host and the parasite and that may be effectively utilized to design specific inhibitors or targets to design antimalarial drugs. Interestingly the first step in the purine salvage pathway differ significantly between the malarial parasites and host. In the malarial parasites, purines formed as products of polyamine synthesis are recycled in a novel pathway in which methyl thioinosine is generated by Adenosine deaminase The enzyme Plasmodium falciparum purine nucleoside phosphorylase converts both 5′- methyl thioinosine and inosine to hypoxanthine and activates the purine pathway that plays an important role in the life cycle of the malarial parasite [6]. We submit an hypothesis on targeting and blocking the protein Adenosine deaminase that will inturn stop the purine pathway of the parasite and may prove to be an effective step in controlling and combating malaria.
Methodology
The sequence of the target protein Adenosine deaminase was obtained from NCBI database [7] and the Protein Data Bank (PDB) [8] was used for obtaining the 3D structure of the desired protein. The tool BLAST was used for searching and finding the correct template homologue of the target sequence [9]. Information about the domain region, region of alignment and function of the domain of the protein was retrieved from Pfam [10].
Homology Modeling:
A bioinformatics tool MODELLER was used for homology modeling of protein 3D structure [11]. A template protein of known 3D structure with enough sequence similarity to the target protein Adenosine deaminase was selected and both the sequences were aligned. Using the alignments the coordinates of the matching residues in the known structure were copied to the unknown protein and the resulting model was evaluated using Combinatorial Extension [12], ProQ server [13] and Ramachandran plot [14].
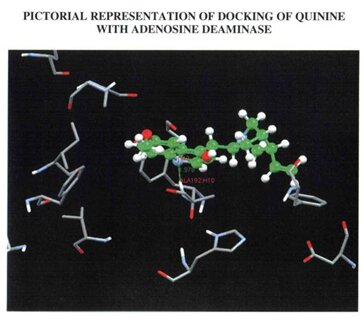
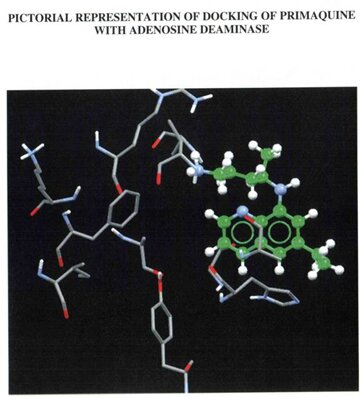
Ligands (drugs) used for targeting the desired protein were drawn using the tool ACD Chemsketch [15]. The sketched structure was then converted to 3D format and optimized to ascertain if the structure obtained was an accurate format to favour docking. Inorder to proceed with the docking studies, the secondary structure of the protein was retrieved from RCSB software through which the active sites, the ligand binding sites and metal binding sites were carefully analyzed. Using another software called WEBLAB [16] hydrogen coordinates were added and docked with the drugs used in this study namely Primaquine (synthetic drug) and Quinine(natural drug). The process of docking was done using a software called GOLD that docks each ligand 10times with the target protein and gives the best result of them.
Results and Discussion
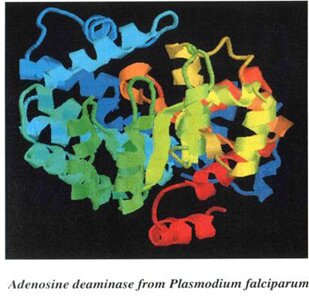
The protein sequence of Adenosine deaminase was retrieved from the target base of NCBI and the template selection was done using PHI-PSI-BLAST based on criterias like, the hit should have the same function, show maximum alignment with the target sequence and the alignment should have less number of gaps. The hit score obtained was 64% which can be considered as a suitable template. A suitable homology model was built using the software MODELLER to retrieve the 3D structure of the target protein that was given as input. The minimum energy level (1790.0635) obtained was the best and the 3D structure was viewed with the help of RASMOL.
The built model was evaluated using Combinatorial Extension (CE), ProQ server and Ramachandran plot. Combinatorial Extension is a database for 3D protein structure comparison and alignment and was used to find out the superimposition of the template and the model. ProQ is a neutral network based predictor and was used to predict the quality of the protein model. Two quality measures LG score and MAX sub were used. Any score greater than 4 was supposed to be a good quality model with regard to LG score and scores greater than 0.5 with regard to MAX sub. The scores obtained for the built model were 6.173 (LG score) and 0.676 (MAX sub). Evaluation of the model using Ramachandran plot showed that the number of residues present in the favoured region is 86.97%, allowed region is 10.76% and outside region is 0.85% . Among the two drugs under study, quinine was found to have an greater GOLD score (43.53) than primaquine (28.86) during docking. Also when the formation of hydrogen bonds between the drugs and their respective targets were studied it showed that the hydroden bond distance between quinine and Adenosine deaminase was 1.979, whereas there was no hydrogen bond established between primaquine and Adenosine deaminase.
Utility
A pioneering approach of merging Bioinformatics and pharmaceutics has been carried out in this hypothesis to study the mechanism of action of drugs in treating life threatening infections like malaria and arrive at a long lasting, safe and effective solution. Keeping in mind the amount of resistance that malarial parasites have developed against conventional drugs, we have proposed a new method of drug designing and target identification based on the purine pathway of the malarial parasite and we have also compared the efficacy of a natural drug against the conventional synthetic drug in treating malaria. We believe this approach will go a long way in helping researches all over the world to find a effective and permanent solution in eradicating malaria.
References
- R.W.Snow et al; Nature, (2005) 434 (7030): 214-7 [PMID:15759000]
- I. Mueller et al; Trends Parasitol (2007) 23 (6): 278–83 [PMID:17459775]
- B. Singh et al; Lancet (2004) 363 (9414): 1017–24 [PMID:15051281]
- K. Mendis et al; Am J Trop Med Hyg (2001) 64 (1-2 Suppl): 97–106 [PMID: 11425182]
- Q. Chen et al; Clin. Microbiol. Rev (2000) 13 (3): 439–50 [PMID:10885986]
- L.M Ting et al; J Biol Chem (2005) 280(10):9547-54 [PMID:15576366]
- http://www.ncbi.nlm.nih.gov/
- http://www.rcsb.org/pdb
- http://www.ncbi.nlm.nih.gov/BLAST/
- pfam.sanger.ac.uk/
- www.salilab.org/modeller/
- http://cl.sdsc.edu/
- www.sbc.su.se/
- G.N. Ramachandran, et al; J. Mol. Biol (1963) 7:95 [PMID: 13990617]
- www.acdlabs.com
- www.umass.edu/microbio/rasmol/othersof.htm
PICTORIAL ILLUSTRATIONS OF THE TARGET PROTEIN (Adenosine deaminase) AND ITS DOCKING RESULTS
WITH A NATURAL DRUG AND SYNTHETIC DRUG.